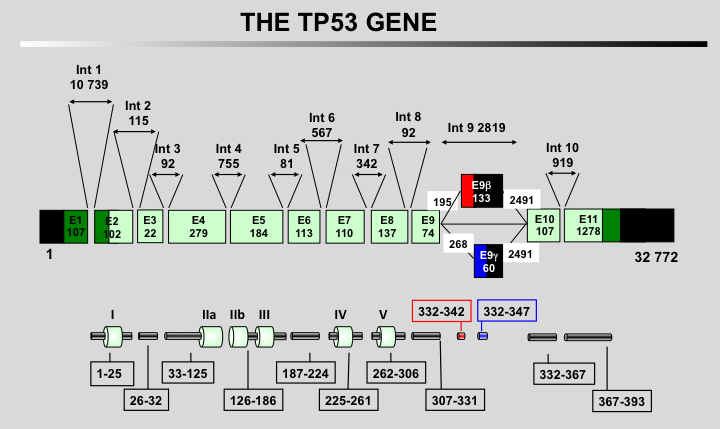
Almost 20 years after the discovery of TP53, two novel homolog genes, TP63 and TP73 were discovered. The most significant homology to TP53 concerns the DNA-binding domain, suggesting some overlapping functions in cell cycle arrest and apoptosis.
The challenge is now to understand the similarities and differences between family members especially in terms of their regulation and potential for physical or genetic interactions with one another.
Click on the links below for further details
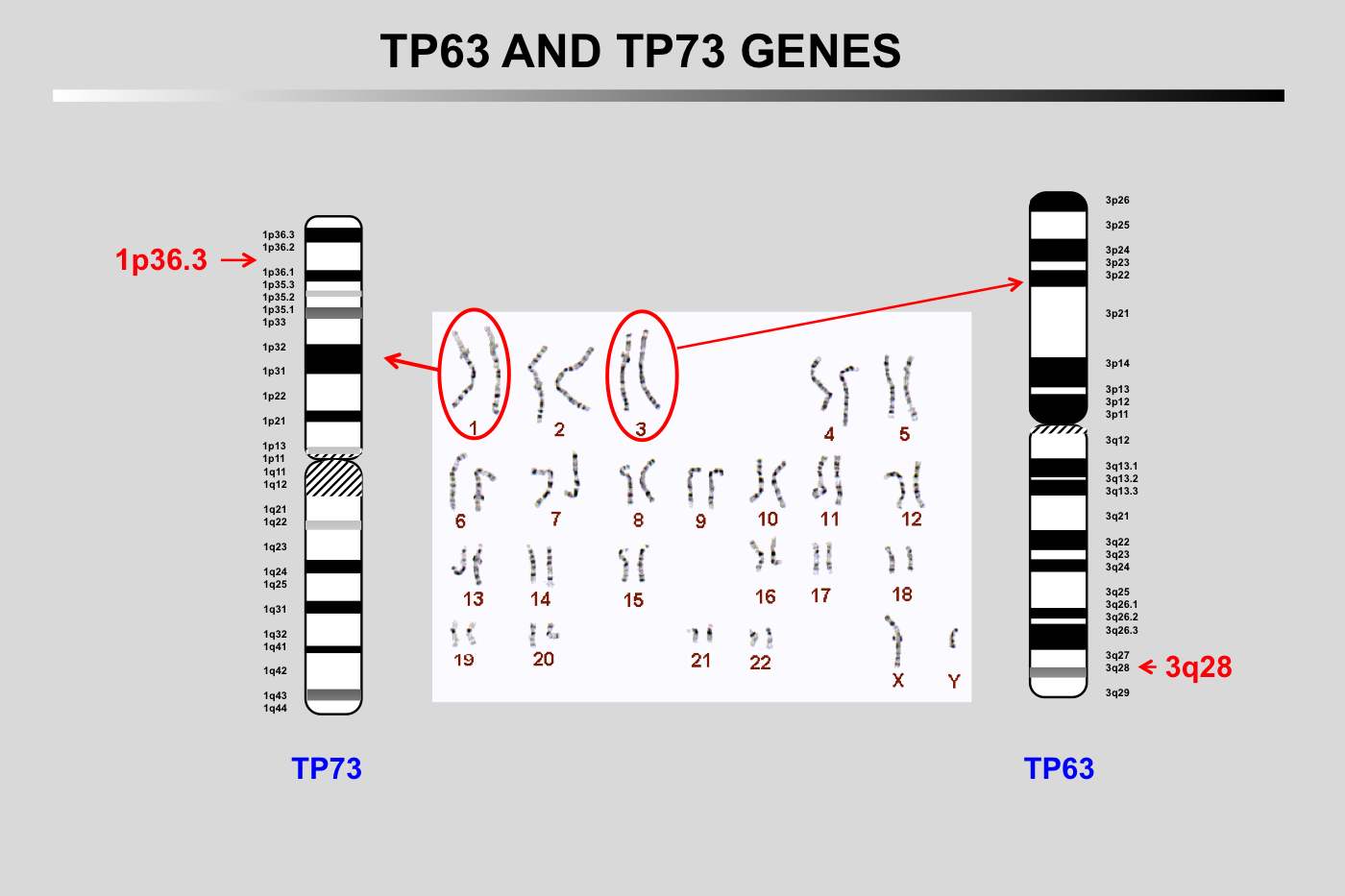
The TP73 gene is localized on chromosome 1p36.3 and has a size of 83,683 bases
The TP63 gene is localized on chromosome 3q28 and has a size of 265,864 bases
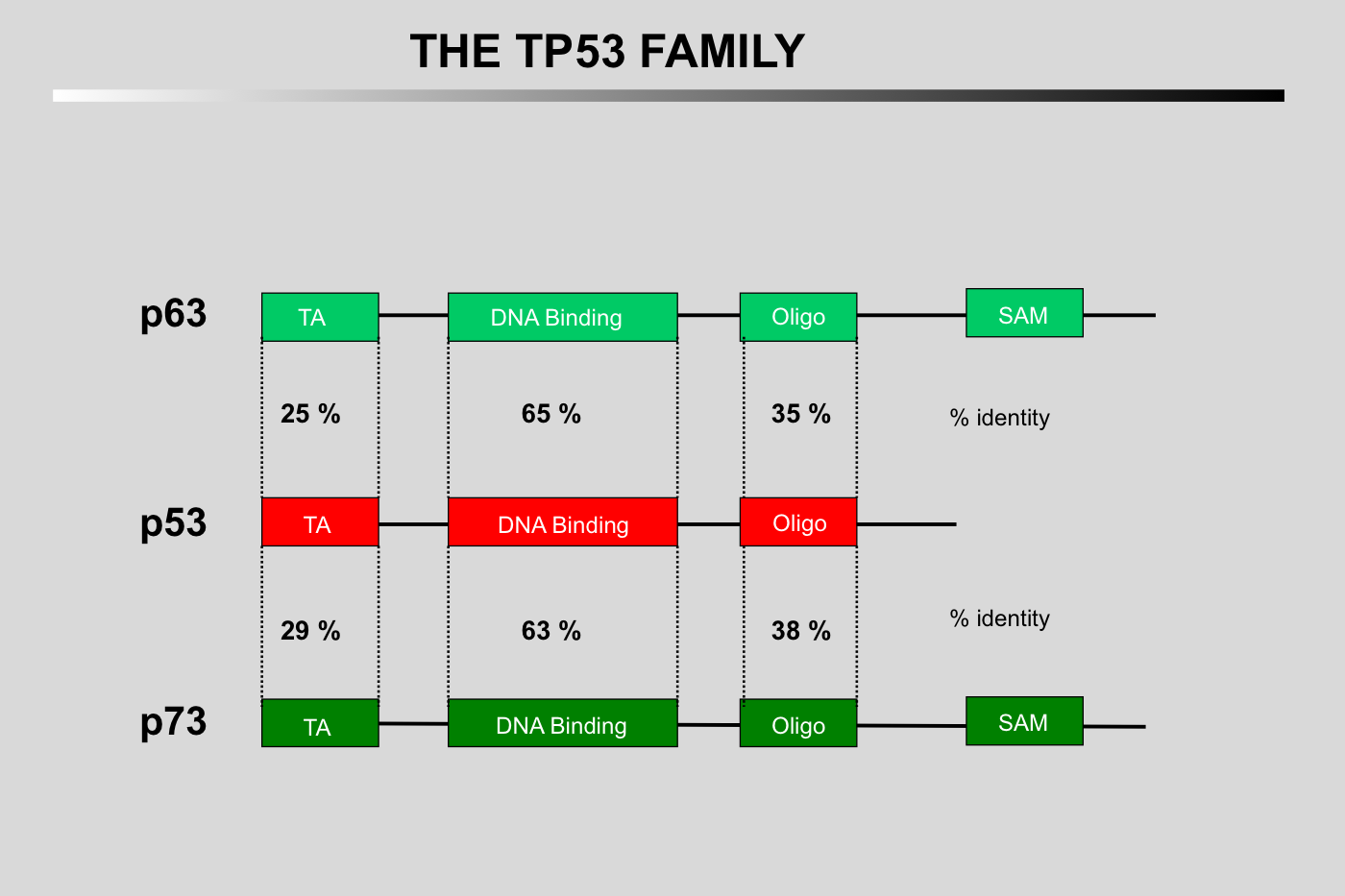
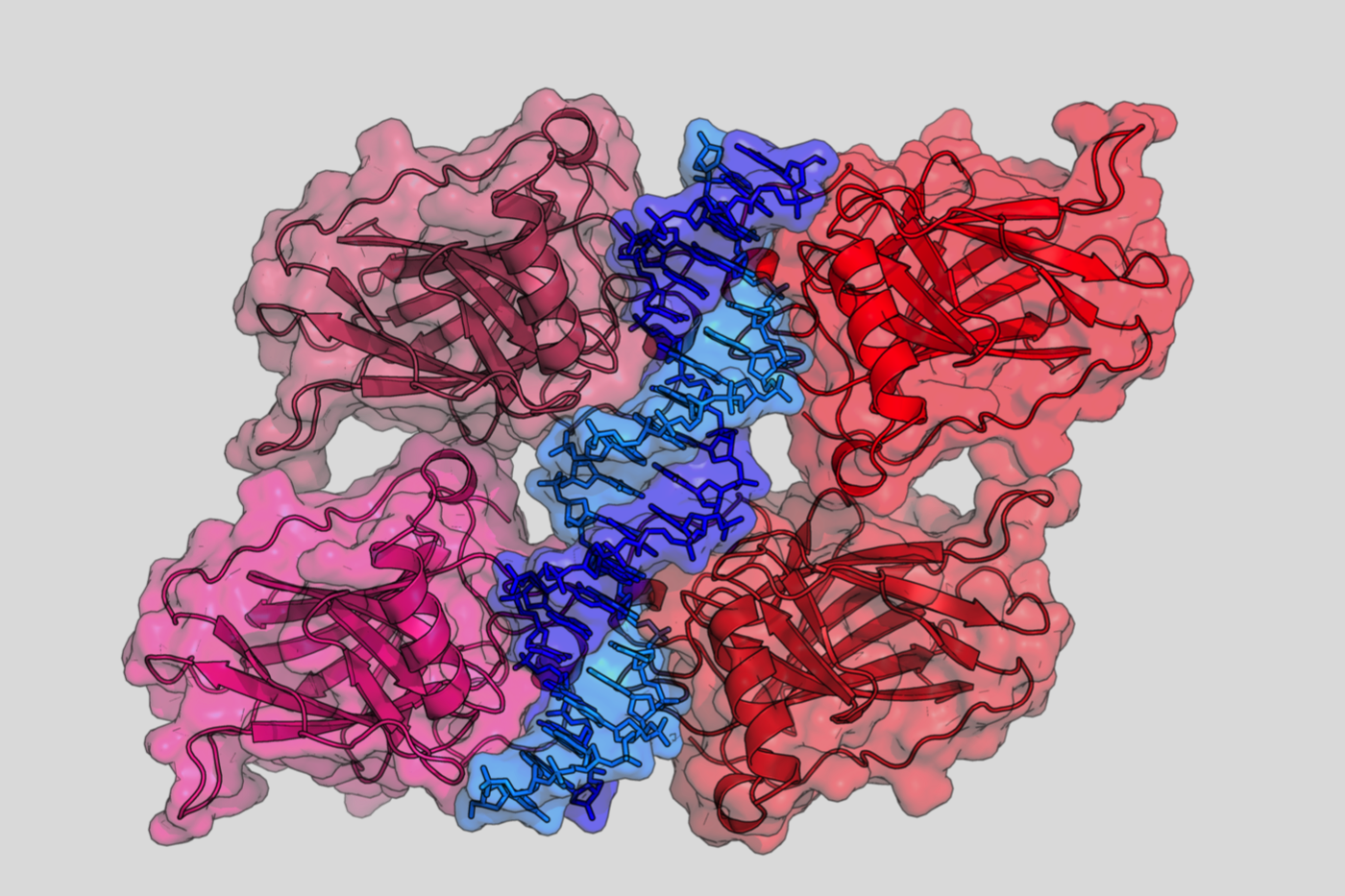
The main homology between the three proteins concerns their DNA-binding domains.
The three proteins can bind similar response elements but with different affinities.
Only the amino-terminus of TP53 can bind to mdm2.
TP73 and TP63 can form hetero-oligomers but cannot oligomerize with the TP53 protein.
The SAM domain is specific to TP73 and TP63.
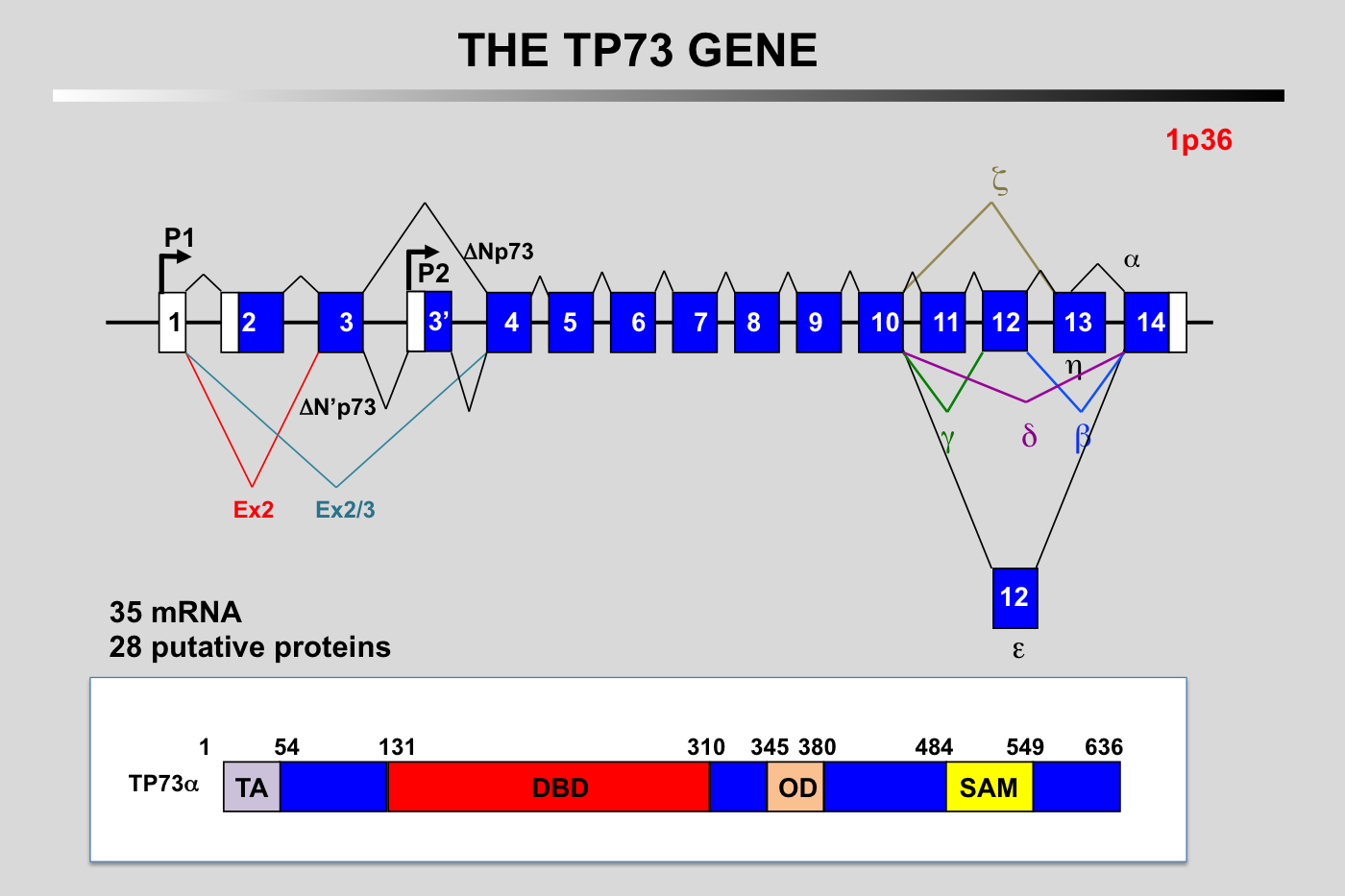
Schematic representation of the exon–intron organization of the human TP73 gene and the alternative splicing events. Alternative promoter use (P1 and P2) gives rise to TA (transactivation) and N-terminally truncated (DN) isoforms. Multiple alternative splicing events will also generate different amino-terminus and at least 5 different carboxy-terminus.
N.B.: the structure of the TP73 gene described above is a synthesis from various published works. Due to the complexity of this gene, it is likely that this picture is not completely accurate.
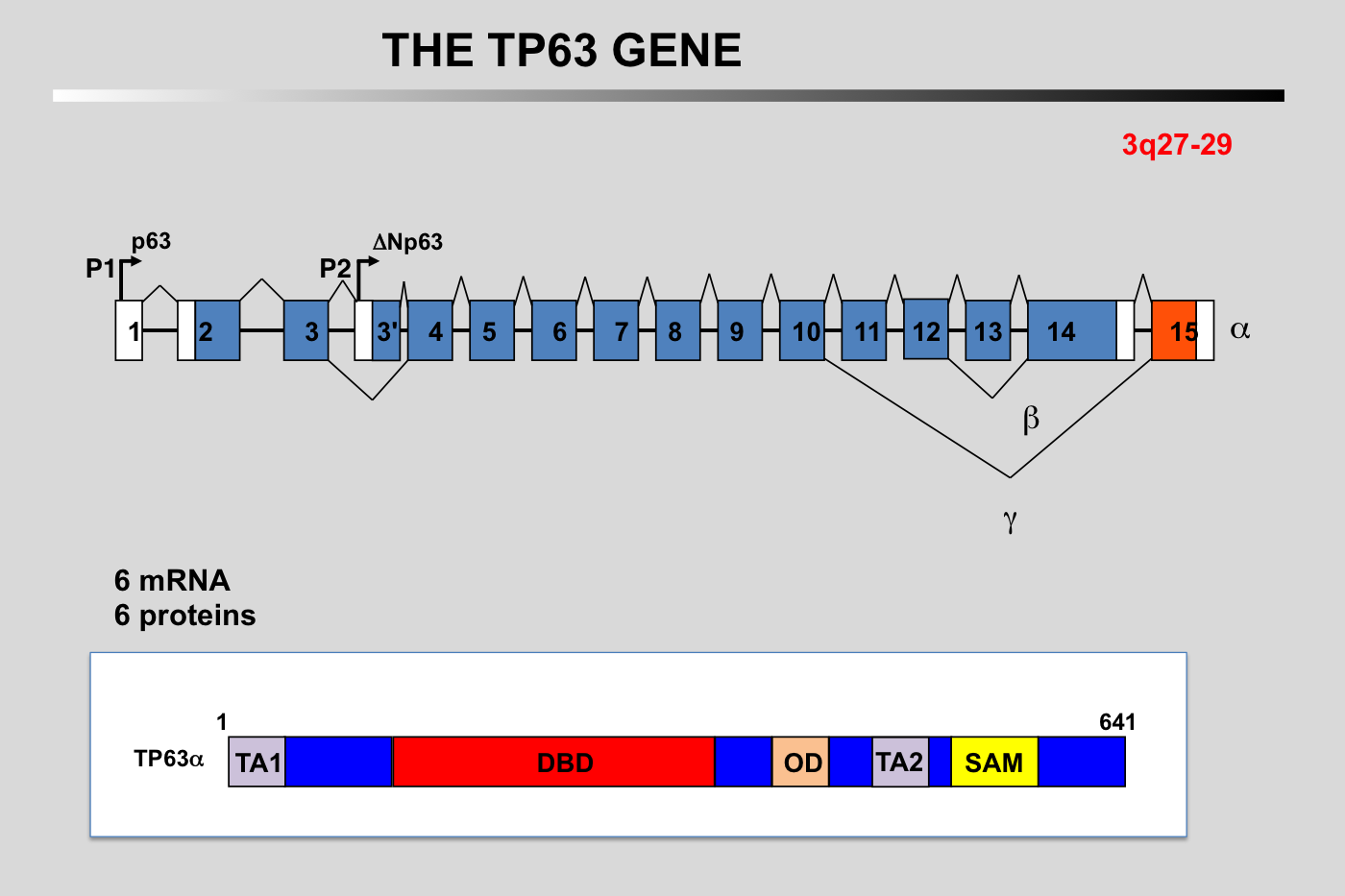
Schematic representation of the exon–intron organization of the human TP63 gene and the alternative splicing events. Alternative promoter use (P1 and P2) gives rise to TA (transactivation) and N-terminally truncated (DN) isoforms. Multiple alternative splicing events will generate at least 5 different carboxy-terminus. The P63 protein comprises up to five types of domains, depending on the isoform, and may contain transactivation (TA1), DNA Binding (DBD), oligomerization (OD), C-terminal sterile alpha motif (SAM), and/or C-terminal transcription inhibitory (ID) domains.
N.B.: the structure of the TP63 gene described above is a synthesis from various published works. Due to the complexity of this gene, it is likely that this picture is not completely accurate.
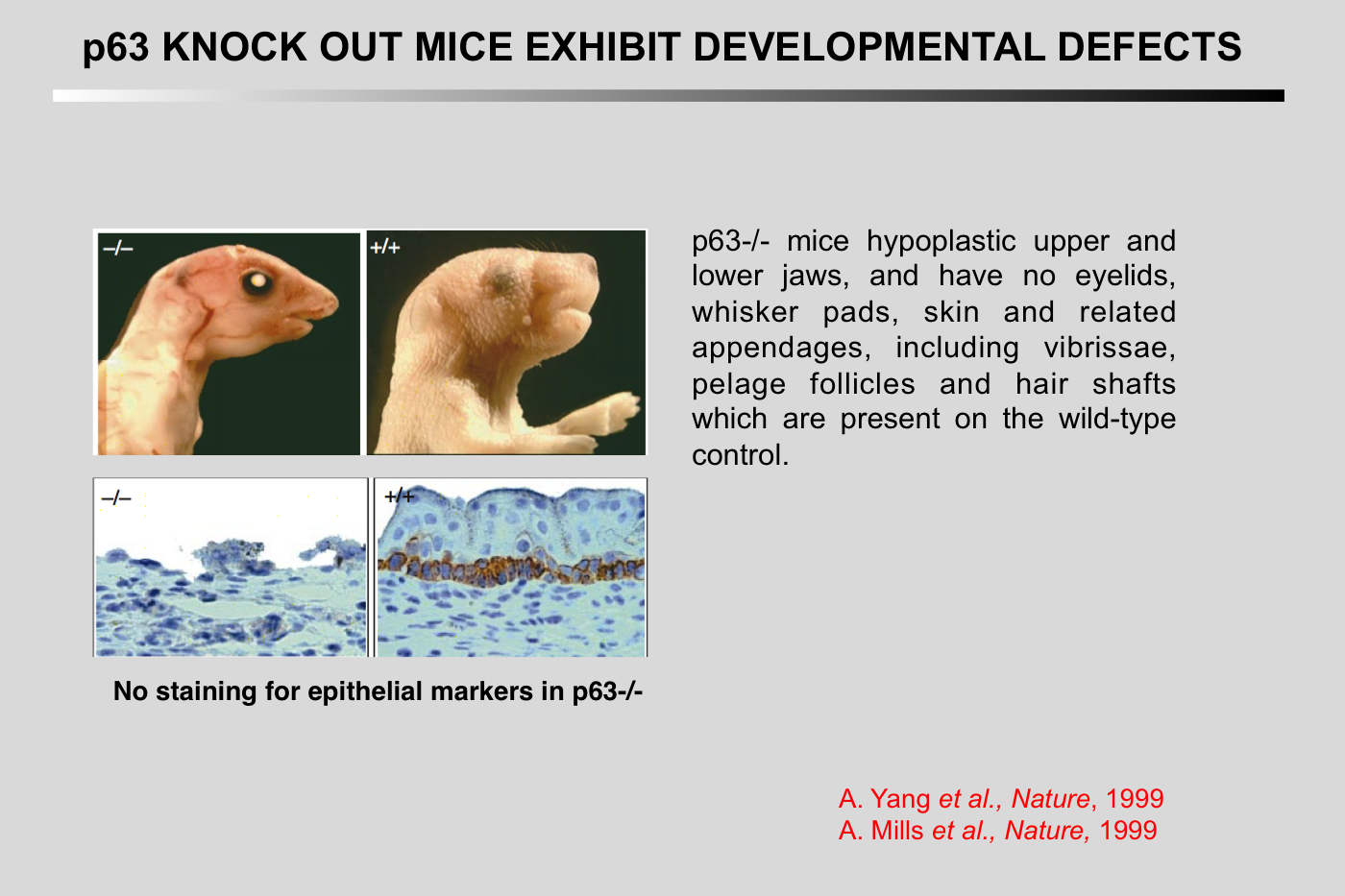
Mills, A. A., B. H. Zheng, X. J. Wang, H. Vogel, D. R. Roop, and A. Bradley (1999) Nature 398:708-713.
p63 is a p53 homologue required for limb and epidermal morphogenesis
Abstract
The p53 tumour suppressor is a transcription factor that regulates the progression of the cell through its cycle and cell death (apoptosis) in response to environmental stimuli such as DNA damage and hypoxia(1,2). Even though p53 modulates these critical cellular processes, mice that lack p53 are developmentally normal(3), suggesting that p53-related proteins might compensate for the functions of p53 during embryogenesis. Two p53 homologues, p63 and p73, are known(4,5) and here we describe the function of p63 in vivo. Mice lacking p63 are born alive but have striking developmental defects. Their limbs are absent or truncated, defects that are caused by a failure of the apical ectodermal ridge to differentiate. The skin of p63- deficient mice does not progress past an early developmental stage: it lacks stratification and does not express differentiation markers. Structures dependent upon epidermal-mesenchymal interactions during embryonic development, such as hair follicles, teeth and mammary glands, are absent in p63-deficient mice. Thus, in contrast to p53, p63 is essential for several aspects of ectodermal differentiation during embryogenesis.
Yang, A., R. Schweitzer, D. Q. Sun, M. Kaghad, N. Walker, R. T. Bronson, C. Tabin, A. Sharpe, D. Caput, C. Crum, and F. McKeon (1999) Nature 398:714-718.
p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development
Abstract
The p63 gene, a homologue of the tumour-suppressor p53 (refs 1-5), is highly expressed in the basal or progenitor layers of many epithelial tissues(1). Here we report that mice homozygous for a disrupted p63 gene have major defects in their limb, craniofacial and epithelial development. P63 is expressed in the ectodermal surfaces of the limb buds, branchial arches and epidermal appendages, which are all sites of reciprocal signalling that direct morphogenetic patterning of the underlying mesoderm. The limb truncations are due to a failure to maintain the apical ectodermal ridge, a stratified epithelium, essential for Limb development The embryonic epidermis of p63(-/-) mice undergoes an unusual process of non-regenerative differentiation, culminating in a striking absence of all squamous epithelia and their derivatives, including mammary, lacrymal and salivary glands. Taken together, our results indicate that p63 is critical for maintaining the progenitor-cell populations that are necessary to sustain epithelial development and morphogenesis.
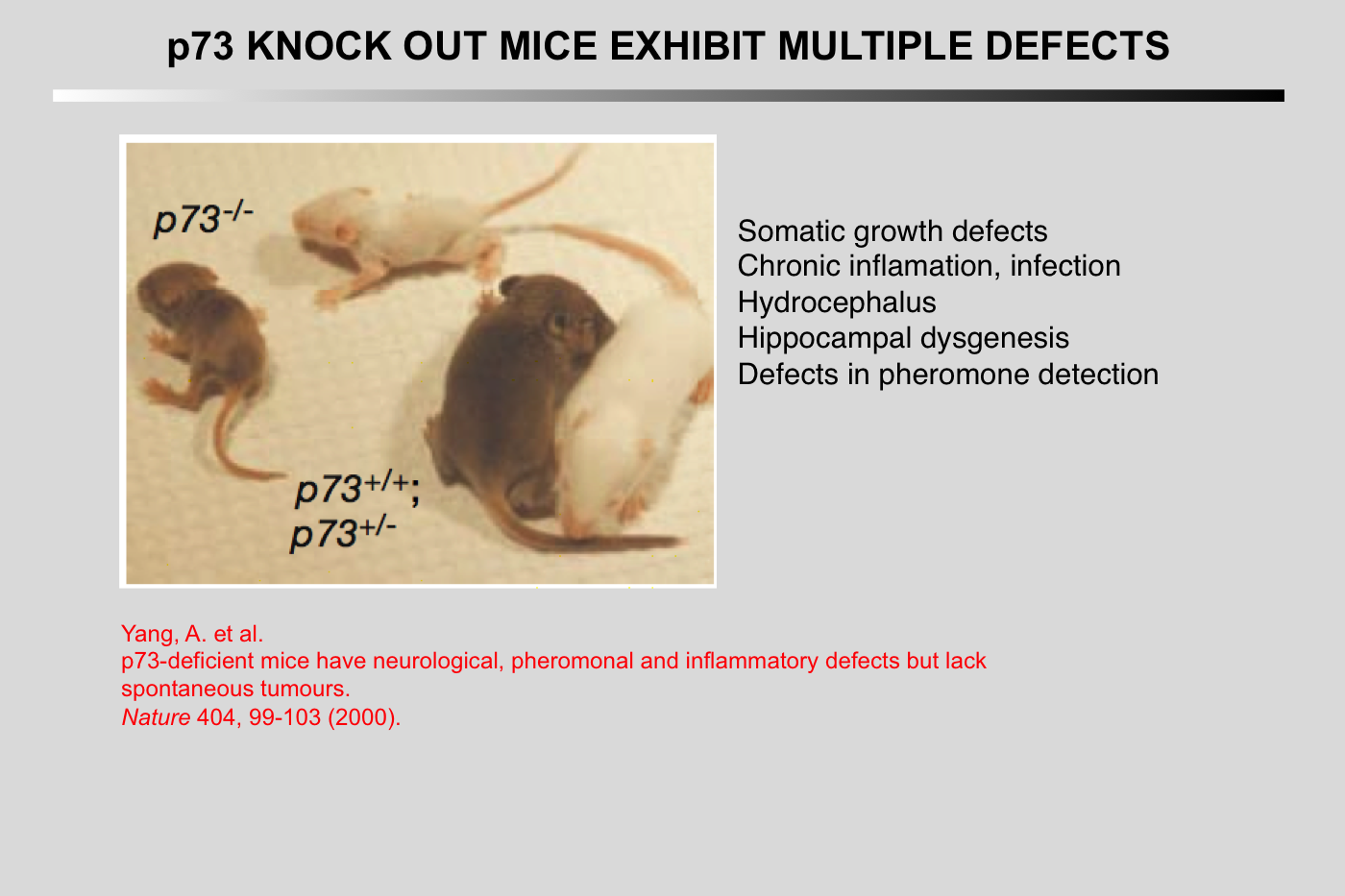
Yang, A., N. Walker, R. Bronson, M. Kaghad, M. Oosterwegel, J. Bonnin, C. Vagner, H. Bonnet, P. Dikkes, A. Sharpe, F. McKeon, and D. Caput (2000) Nature 404:99-103.
p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours
Abstract
p73 (ref. 1) has high homology with the tumour suppressor p53 (refs 2- 4), as well as with p63, a gene implicated in the maintenance of epithelial stem cells. Despite the localization of the p73 gene to chromosome 1p36.3, a region of frequent aberration in a wide range of human cancers, and the ability of p73 to transactivate p53 target genes, it is unclear whether p73 functions as a tumour suppressor. Here we show that mice functionally deficient for all p73 isoforms exhibit profound defects, including hippocampal dysgenesis, hydrocephalus, chronic infections and inflammation, as well as abnormalities in pheromone sensory pathways. In contrast to p53-deficient mice, however, those lacking p73 show no increased susceptibility to spontaneous tumorigenesis. We report the mechanistic basis of the hippocampal dysgenesis and the loss of pheromone responses, and show that new, potentially dominant-negative, p73 variants are the predominant expression products of this gene in developing and adult tissues. Our data suggest that there is a marked divergence in the physiological functions of the p53 family members, and reveal unique roles for p73 in neurogenesis, sensory pathways and homeostatic control.
Heterozygous mutations in the transcription factor gene T63 are causative for several developmental syndromes with ectodermal dysplasia, orofacial clefting and limb malformations as the key characteristics.
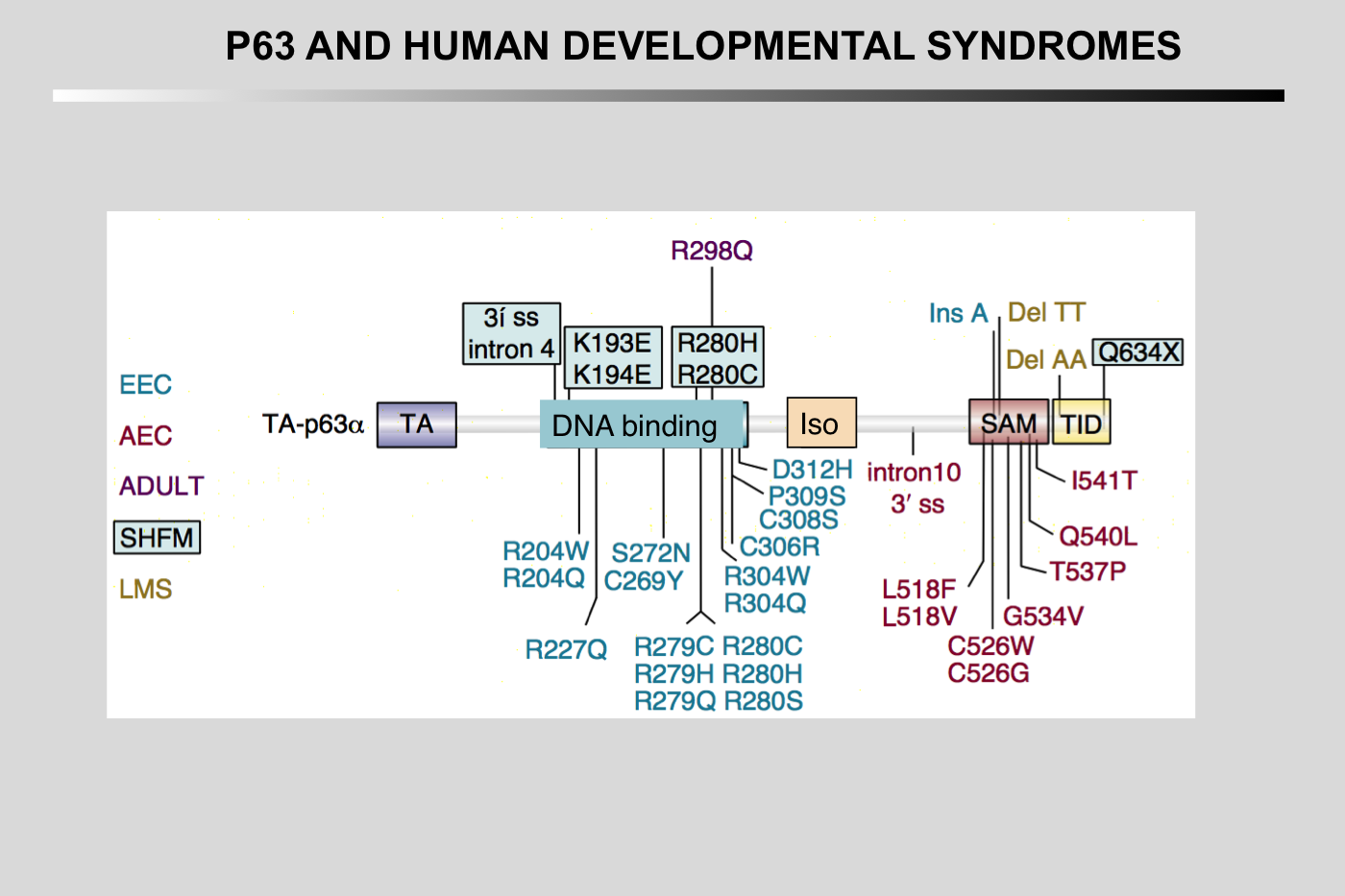
Pathogenic p63 mutations cause at least five different syndromes and two non-syndromic conditions. Mutations causing different diseases are illustrated in different colors. Only mutations that are discussed in the text are indicated, an overview of all mutations is given in Table 1. EEC hotspot mutations are clustered in DNA-binding domain, and RHS and AEC syndrome mutations in SAM and TI domains. Several mutations, such as R280, R313, I510, S541 and 1850 Del A, can have a variable clinical outcome, probably due to genetic background effects. The black asterisks illustrate sites needed for ubiquitination (K193, K194 and PY) and the white asterisk represents a sumoylation site (φKXD/E).
EEC: ectrodactyly, ectodermal dysplasia, clefting
LMS: limb-mammary syndrome
ADULT: acro-dermato-ungual-lacrimal-tooth
AEC: ankyloblepharon, ectodermal dysplasia, clefting
RHS: Rapp- Hodgkin syndrome
SHFM: split hand/foot malformation
NSCL: non-syndromic cleft lip.
Note the clustering of mutations in the DNA-binding domain for ectrodactyly-ectodermal dysplasia-clefting (EEC) syndrome and in the sterile α motif (SAM) domain for ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome.
Figure from Rinne T, Brunner HG, van Bokhoven H (2007) p63-associated disorders. Cell Cycle 6: 262-268. 17224651
Costanzo A, Pediconi N, Narcisi A, Guerrieri F, Belloni L, Fausti F, Botti E, Levrero M (2014) TP63 and TP73 in cancer, an unresolved „family" puzzle of complexity, redundancy and hierarchy. FEBS Lett 588(16): 2590-2599.
Candi E, Agostini M, Melino G, Bernassola F (2014) How the TP53 Family Proteins TP63 and TP73 Contribute to Tumorigenesis: Regulators and Effectors. Hum Mutat 35: 702-714.
Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH (2013) Metabolic Regulation by p53 Family Members. Cell Metab 18: 617-633.
Flores ER, Lozano G (2012) The p53 family grows old. Genes Dev 26: 1997-2000.
Levine AJ (2012) The evolution of the p53 family of genes. Cell Cycle 11: 214-215.
Wei J, Zaika E, Zaika A (2012) p53 Family: Role of Protein Isoforms in Human Cancer. J Nucleic Acids 2012: 687359.
Candi E, Agostini M, Melino G, Bernassola F (2014) How the TP53 Family Proteins TP63 and TP73 Contribute to Tumorigenesis: Regulators and Effectors. Hum Mutat 35: 702-714