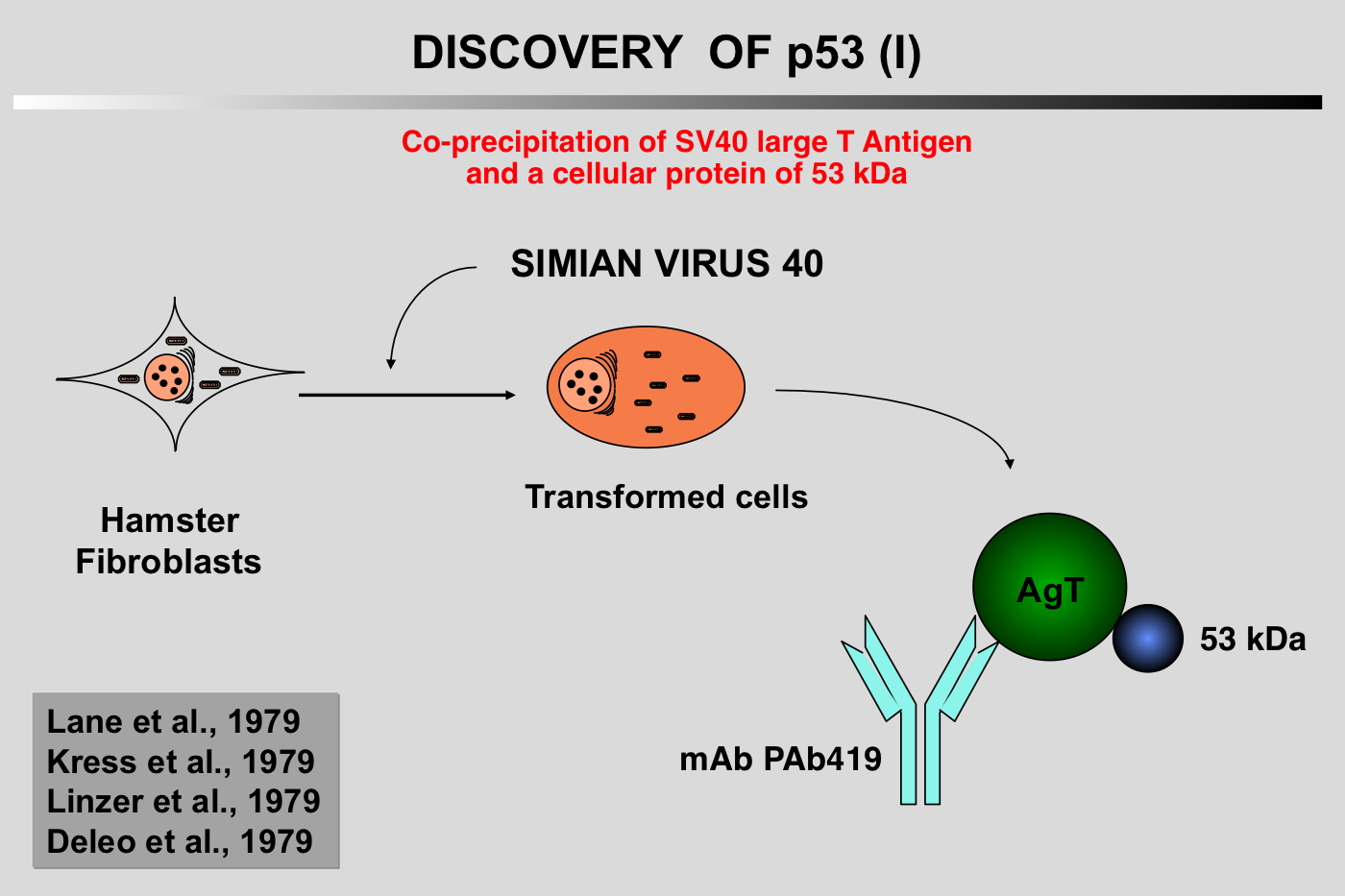
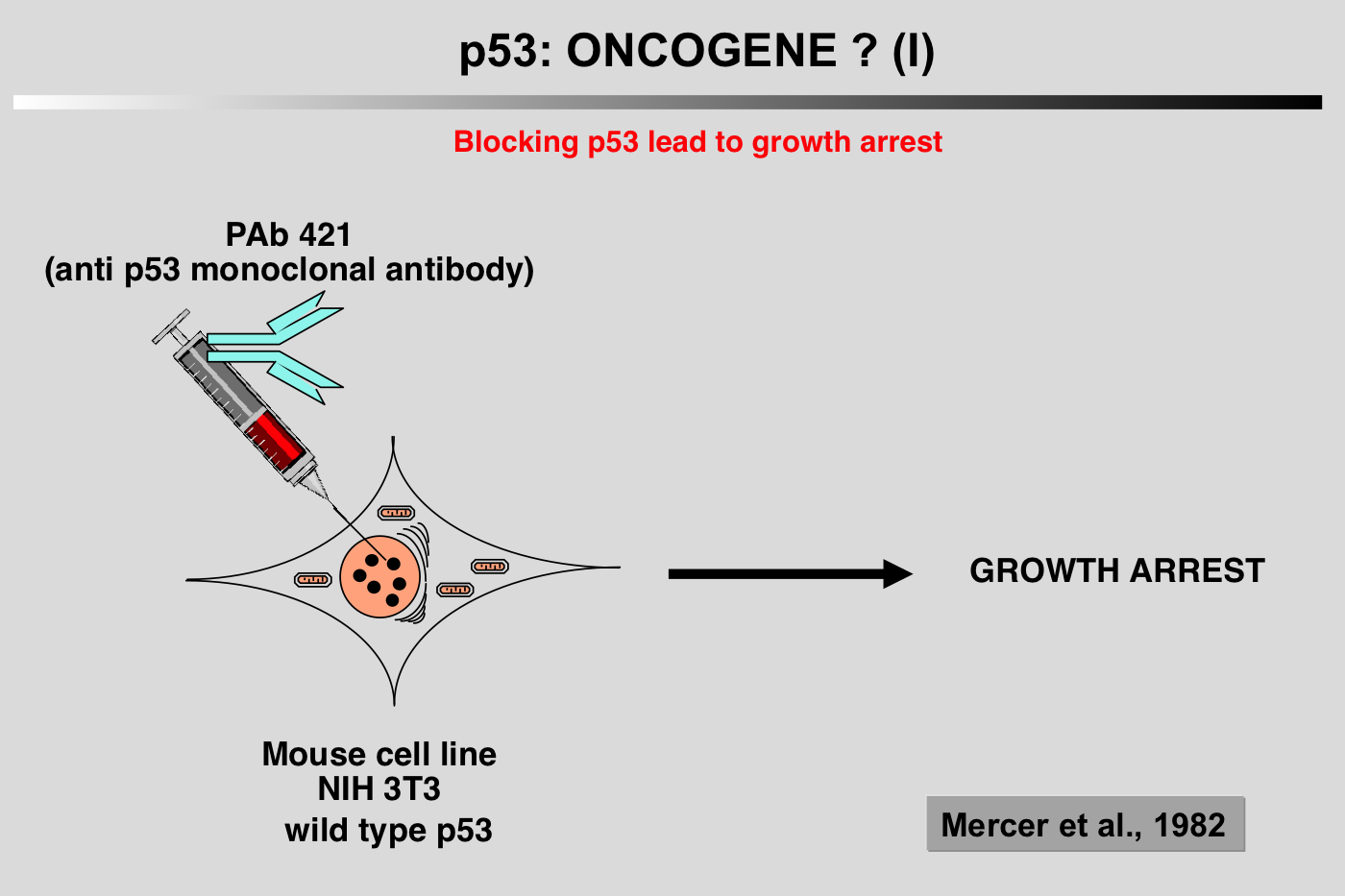
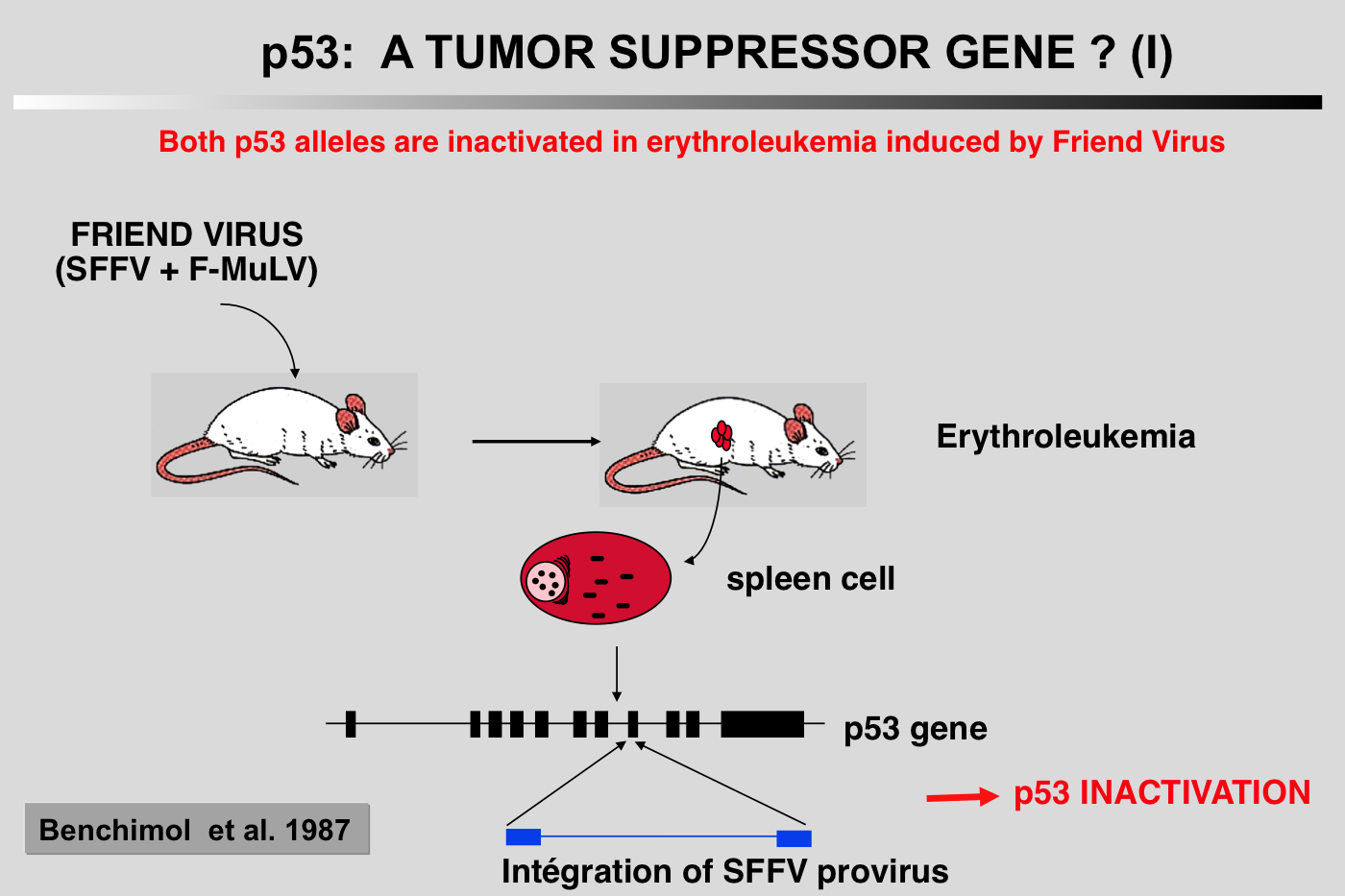
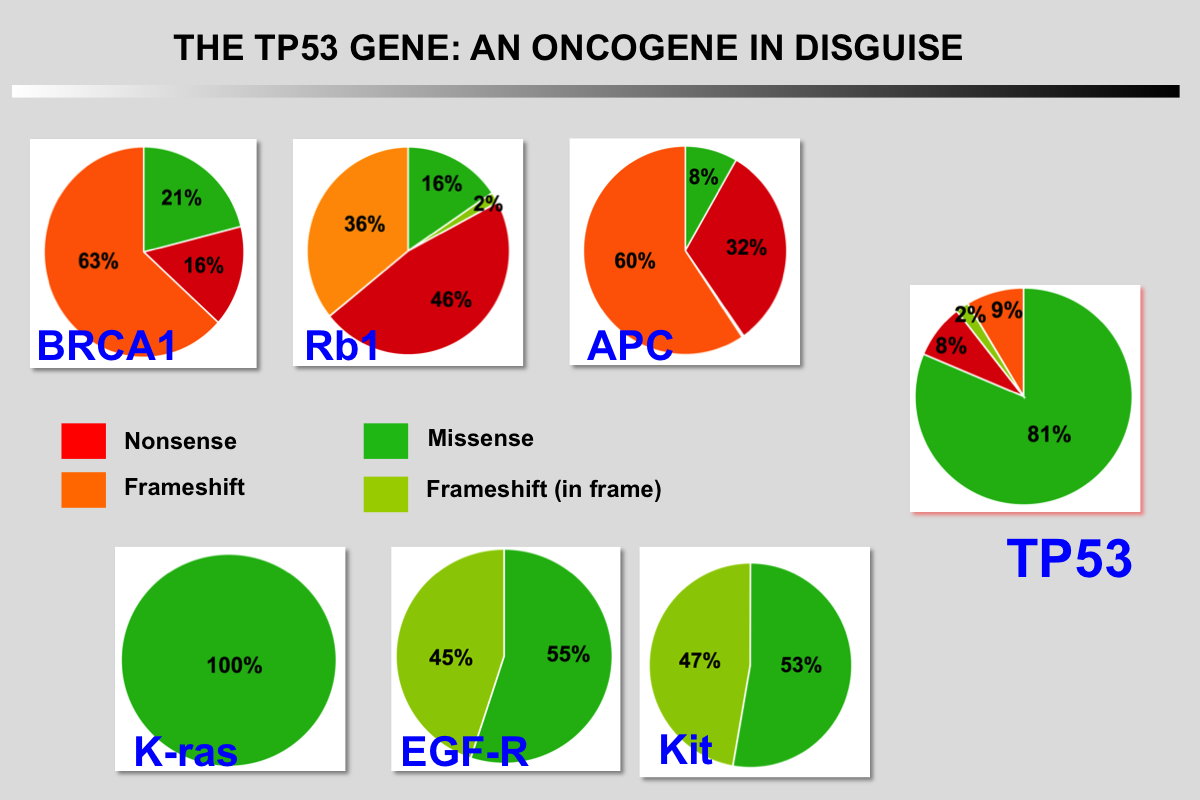
|closedFrom 1985 to 1988, p53 research started to encounter a number of difficulties and the 131 articles published on the topic during this period did not include p53 in the family of oncogenes.
One of these studies showed that both p53 alleles were inactivated in murine erythroleukemia induced by the Friend virus (Mowat et al, 1985). Another 'strange' observation was the high frequency of p53 gene rearrangements and deletions in human and murine osteosarcoma (Masuda et al, 1987; Chandar et al, 1992.
Click on the links below for further details
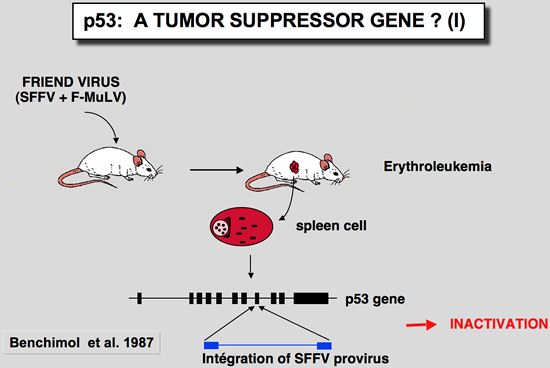
In these tumors induced by the Friend virus, the p53 gene found in tumor cells was very often rearranged, leading to absence of expression or synthesis of a truncated or mutant protein (Mowat et al. 1985). The mutation often affects one of the conserved blocks of the protein (Munroe et al. 1988). In all cases studied, the second allele is either lost through loss of the chromosome, or inactivated by deletion. In this tumor model, functional inactivation of the p53 gene seems to confer a selective growth advantage to erythroid cells during the development of Friend leukemia in vivo.
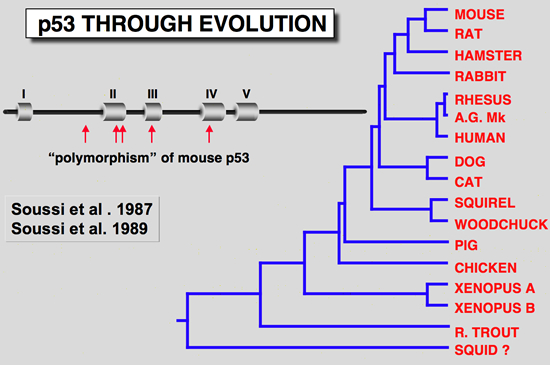
The finding that one murine p53 cDNA clone isolated from the F9 cell failed to cooperate with an activated Ha-ras gene was another clue that the p53 cDNA clones differ from one another in terms of their behavior (Finlay et al. 1988). Examination of all available murine p53 cDNA clones revealed several codon changes which were primarily assumed to be due to polymorphism. However, comparison of these sequence differences with p53 from lower species indicated that some of them occurred in highly conserved regions (Soussi et al. 1990; Soussi et al. 1987), a feature which is not linked to polymorphism. Careful reinvestigation of all sequences led to the conclusion that the F9 cDNA clone was a wild-type, while most of the others used in transfection experiments contained point mutations which activate their transforming properties.
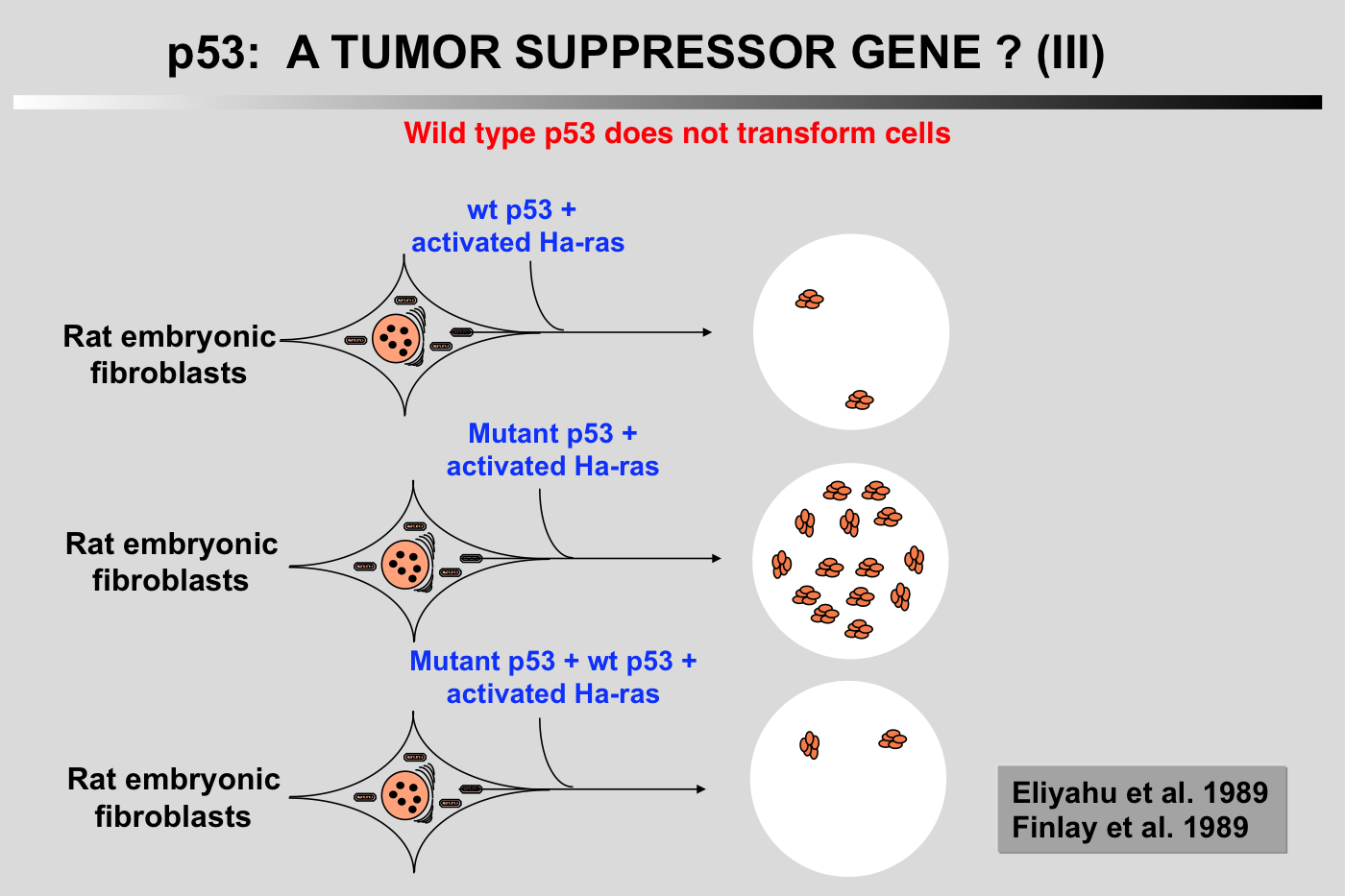
Wild-type p53 has antiproliferative properties and does not cooperate with Ha-ras: A new set of experiments has shown that cotransfection of a plasmid encoding wild-type p53 reduced the transformation potential of plasmids encoding p53 and an activated Ha-ras gene (Eliyahu et al. 1989; Finlay et al. 1989). Furthermore, wild-type p53 was shown to suppress transformation by a mixture of E1A or myc and an activated Ha-ras gene. These transformation experiments indicated that wild-type p53 is a suppressor of cell transformation in vitro.
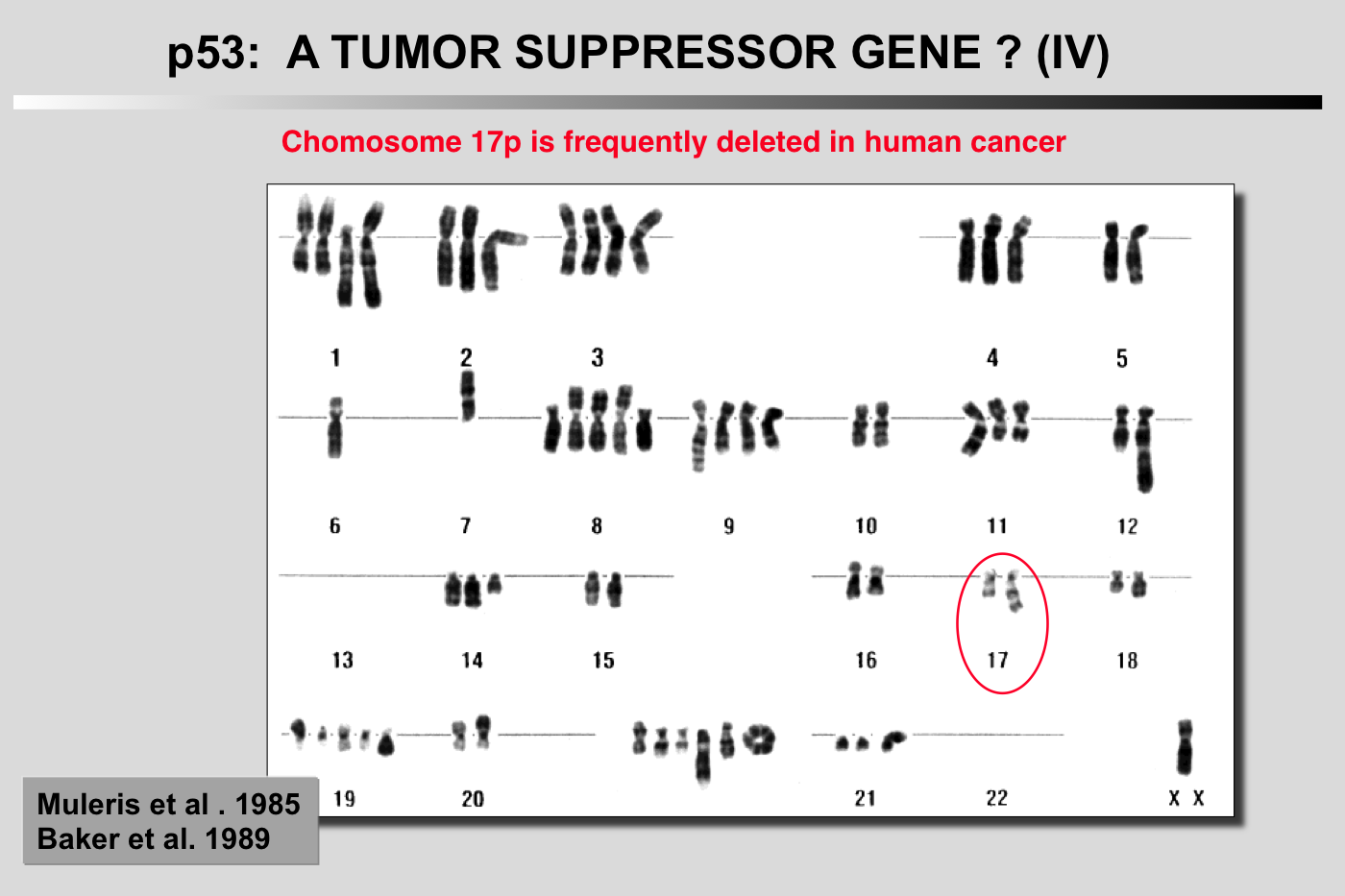
Cytogenetic study of 11 cases of colorectal carcinoma was performed after R-banding. In all instances, there was a rearrangement involving chromosome 17 in its juxtacentromeric region, leading to loss of its short arm. There was also a relative lack of chromosome 18, unrelated to a rearrangement of this chromosome in all but one case. Other anomalies, involving chromosomes 1 and 8 among others, were frequently but not systematically observed. The consistent lack of chromosome 18 and of the short arm of chromosome 17, leading to complete or partial monosomy of these chromosomes in near-diploid cells, suggests that the passage to the hemizygous status of recessive genes carried by these chromosomes may play an important role in the development of colorectal carcinoma.
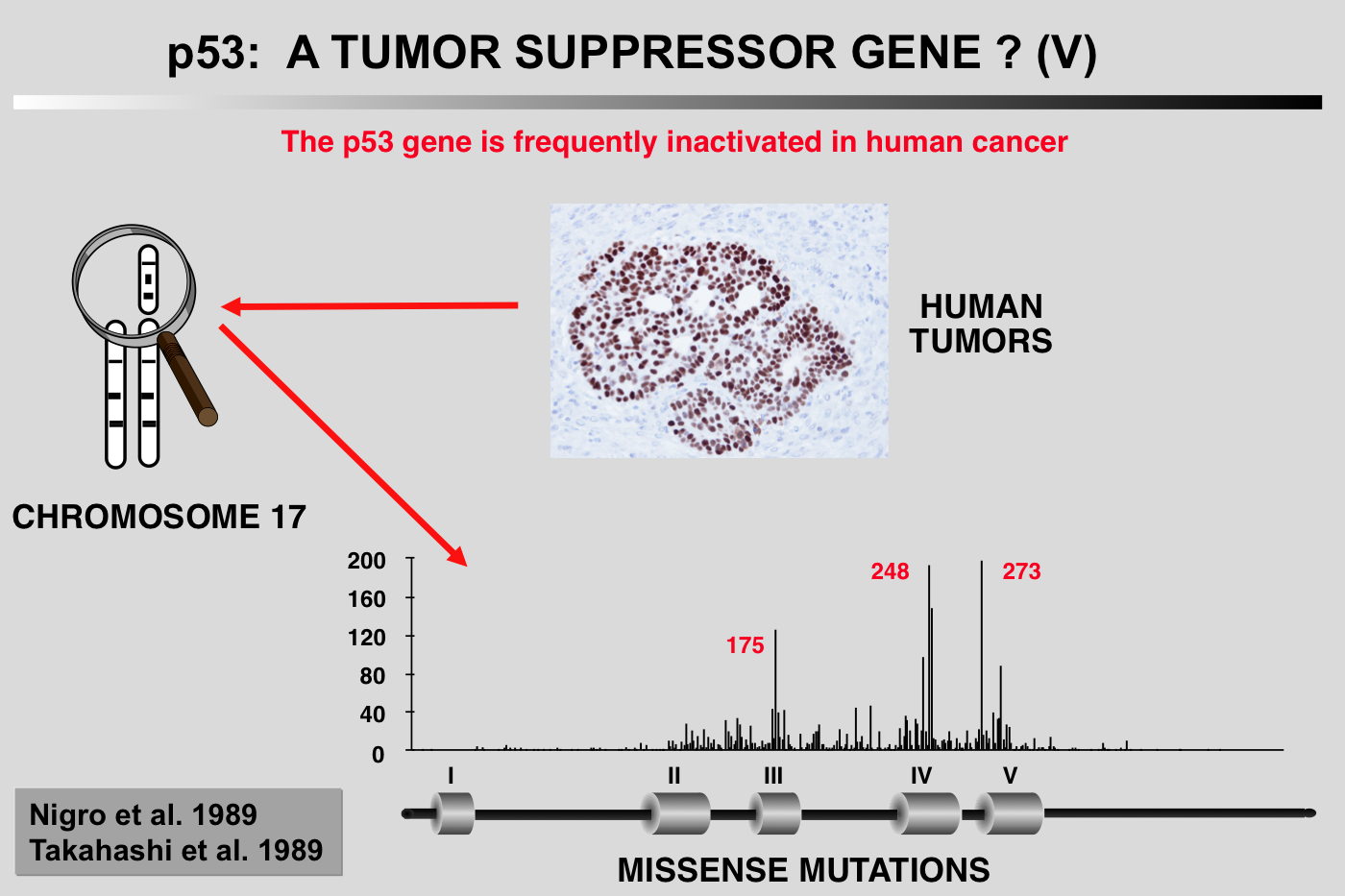
The expression of p53 in various human cancers or tumor cell lines has been studied for many years by several different investigators. p53 expression is often high, but no precise explanation for this phenomenon is available, as, apart from a few cases of osteosarcoma, no gene rearrangements, detectable by Southern blotting, have been identified. Genetic analysis of colorectal cancer reveals a very high rate of heterozygous loss of the short arm of chromosome 17, which carries the p53 gene (Vogelstein et al. 1988).
PCR analysis and sequencing of the remaining p53 allele show that it often contains a point mutation (Baker et al. 1989). Similar observations have been reported in the case of lung cancer (Takahashi et al. 1989). On the heels of these initial observations have come several hundred reports of alterations of the p53 gene in all types of human cancer (see below). In many cases, these mutations are accompanied by heterozygous loss of the short arm of chromosome 17.
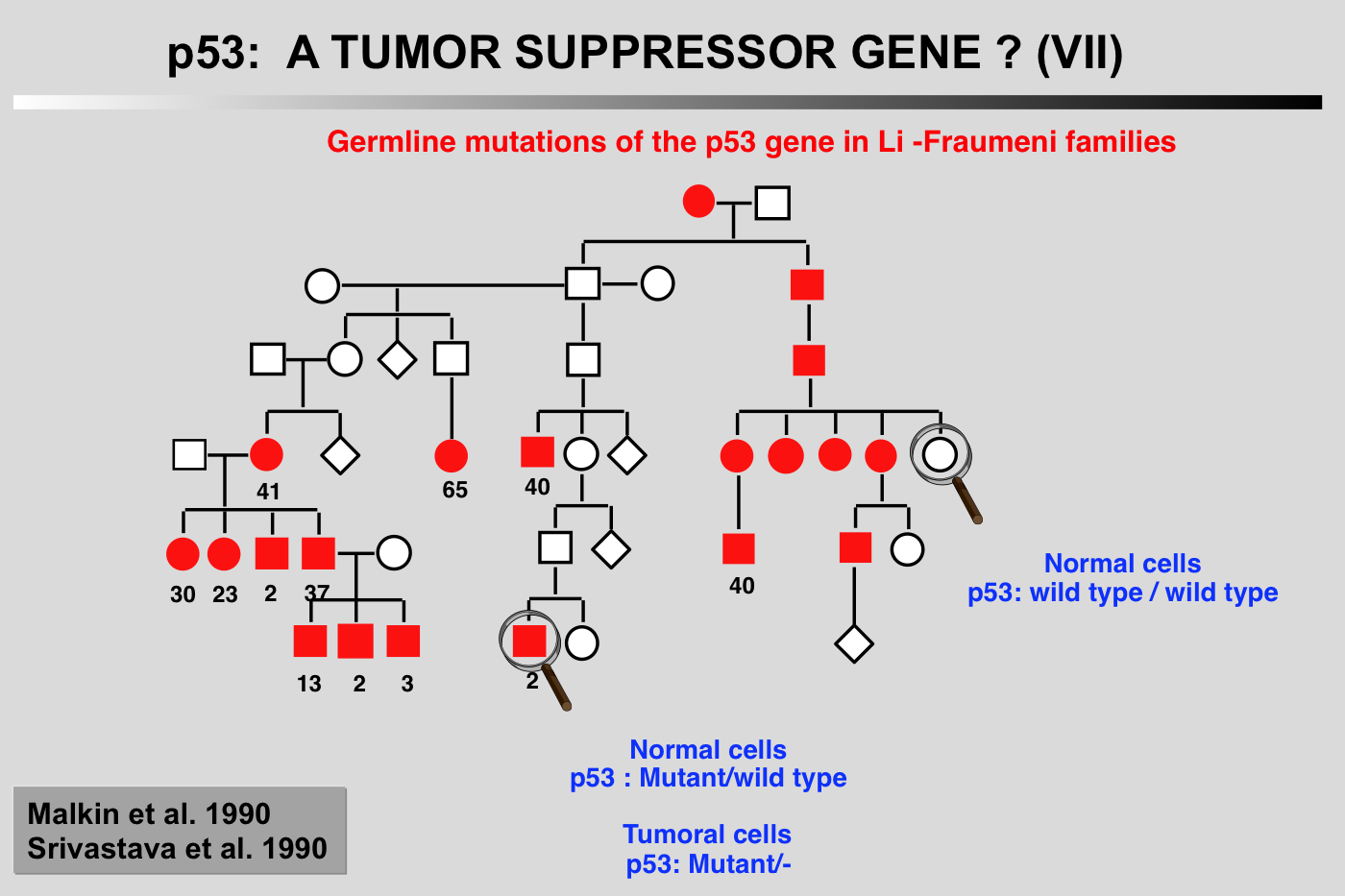
Li-Fraumeni syndrome presents as a familial association of a broad spectrum of cancers including osteosarcomas, breast cancer, soft tissue sarcoma and leukemia, appearing at a very early age. Statistical analysis predicts that 50% of these individuals will develop a tumor before the age of 30, and 90% before the age of 70. Germ-line mutations in the p53 gene have been found in several families with this syndrome (Malkin et al. 1990; Srivastava et al. 1990). In all cases, strict correlation is observed between transmission of the mutant allele and development of a cancer.
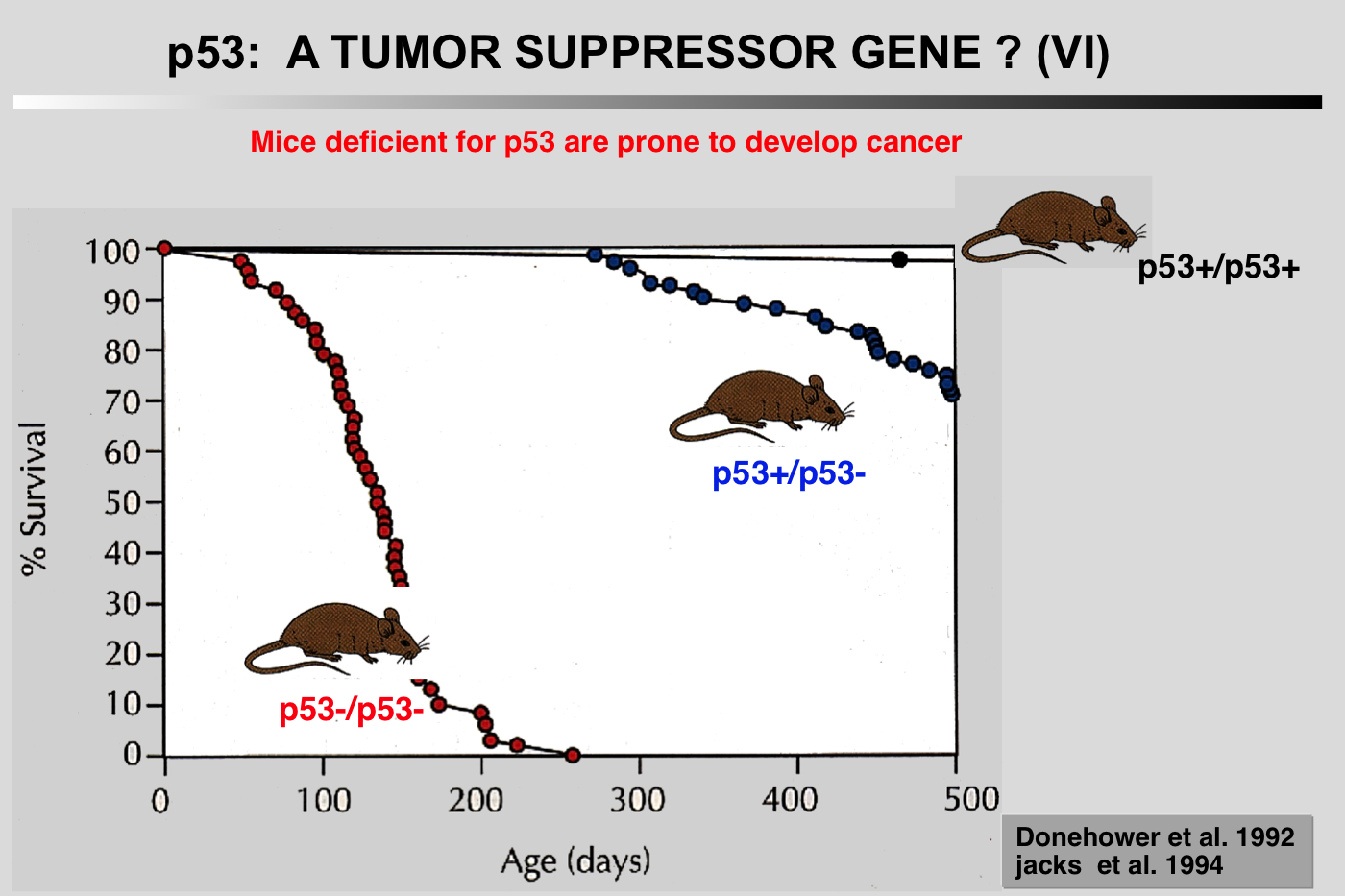
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Butel JS and Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 356: 215-221.
Abstract of the paper
Mutations in the p53 tumor-suppressor gene are the most frequently observed genetic lesions in human cancers. To investigate the role of the p53 gene in mammalian development and tumorigenesis, a null mutation was introduced into the gene by homologous recombination in murine embryonic stem cells. Mice homozygous for the null allele appear normal but are prone to the spontaneous development of a variety of neoplasms by 6 months of age. These observations indicate that a normal p53 gene is dispensable for embryonic development, that its absence predisposes the animal to neoplastic disease, and that an oncogenic mutant form of p53 is not obligatory for the genesis of many types of tumors
The notion that mice null for the TP53 gene are normal except for their high propensity to develop cancer is a misconception. Absence of TP53 leads to developmental defects as well as poor fertility in female mice, a phenotype that varies considerably among the various mouse strains. Whether this phenotype is related to the tumor suppressive function of TP53 remains to be defined.
References
Baker SJ, Fearon ER, Nigro J, Hamilton S, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, Whyte R and Vogelstein B (1989) Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science 244: 217-221.
Eliyahu D, Michalovitz D, Eliyahu S, Pinhasikimhi O and Oren M (1989) Wild-Type p53 can inhibit oncogene-mediated focus formation. Proc. Natl. Acad. Sci. USA 86: 8763-8767.
Finlay CA, Hinds PW and Levine AJ (1989) The p53 proto-oncogene can act as a suppressor of transformation. Cell 57: 1083-1093.
Finlay CA, Hinds PW, Tan TH, Eliyahu D, Oren M and Levine AJ (1988) Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half life. J. Virol. 8: 531-539.
Halevy O, Michalovitz D and Oren M (1990) Different tumor-derived p53 mutants exhibit distinct biological activities. Science 250: 113-116.
Lavigueur A, Maltby V, Mock D, Rossant J, Pawson T and Bernstein A (1989) High incidence of lung, bone, and lymphoid tumors in transgenic mice overexpressing mutant alleles of the p53 oncogene. Mol. Cell. Biol. 9: 3982-3991.
Malkin D, Li FP, Strong LC, Fraumeni JF, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA and Friend SH (1990) Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250: 1233-1238.
Michalovitz D, Halevy O and Oren M (1991) p53 mutations - gains or losses. J. Cell. Biochem. 45: 22-29.
Milner J and Medcalf EA (1991) Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65: 765-774.
Mowat M, Cheng A, Kimura N, Bernstein A and Benchimol S (1985) Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 314: 633-636.
Munroe DG, Rovinski B, Bernstein A and Benchimol S (1988) Loss of highly conserved domain on p53 as a result of gene deletion during friend virus-induced erythroleukemia. Oncogene 2: 621-624.
Soussi T, Caron de Fromentel C and May P (1990) Structural aspects of the p53 protein in relation to gene evolution. Oncogene 5: 945-952.
Soussi T, Caron de Fromentel C, Méchali M, May P and Kress M (1987) Cloning and characterization of a cDNA from Xenopus laevis coding for a protein homologous to human and murine p53. Oncogene 1: 71-78.
Srivastava S, Zou ZQ, Pirollo K, Blattner W and Chang EH (1990) Germ-line transmission of a mutated p53 gene in a cancer-prone family with li-fraumeni syndrome. Nature 348: 747-749.
Takahashi T, Nau MM, Chiba I, Birrer MJ, Rosenberg RK, Vinocour M, Levitt M, Pass H, Gazdar AF and Minna JD (1989) p53 - a frequent target for genetic abnormalities in lung cancer. Science 246: 491-494.
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nacamura V, White R, Smits AM and Bos JL (1988) Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319: 525-532.